Учебные материалы
Конектбиофарм
Работа
Компании
Реклама от Google
Несовершенный остеогенез
Категории: Костная патология, Метаболические заболевания костей,
Несовершенный остеогенез (osteogenesis imperfecta) — врожденная ломкость костей. Это сложное заболевание костей и некоторых соединительнотканных структур, имеющее широчайший диапазон изменений, известно с глубокой древности как заболевание с выраженной клинической картиной и различными формами, передающееся по наследству. Первые упоминания о нем появились в XVII в. В конце XVIII в., т.е. 200 лет назад, Olaus Jacob Ekmann описал НО у членов одной семьи, N. Ekroth (1788) сообщил о заболевании, которое в четырех семьях передавалось детям, и назвал его osteomalacia congenita. Axmann (1831) не только описал ломкость костей у себя и брата, но и, очевидно, первый отметил такой важный симптом, как наличие голубых склер.
Lobstein (1833) описал ломкость костей у больных различного возраста. По данным Vrolik (1849), переломы у детей происходили или еще внутриутробно, или вскоре после рождения. Е. Looser (1906) описал эти две формы как osteogenesis imperfecta congenita und tarda.
Изучением заболевания занимались многие врачи, описавшие более 20 различных симптомов, из которых основными являются:
изменения в строении скелета и легко наступающие переломы, часто небольшой рост; голубые склеры; опаловидный дентин (dentinogenesis imperfecta); прогрессирующая деформация позвоночника, грудной клетки, черепа и длинных трубчатых костей; тугоухость по проводниковому типу; гиперэкстензия в суставах и их деформация; изменения со стороны сердца и крупных сосудов, носовые кровотечения и др.
Работами последних лет показано, что несовершенный остеогенез является гетерогенным наследственным заболеванием генетической природы, поражающим соединительную ткань и выражающимся остеопенией и вышеперечисленными клиническими признаками.
Вместо двух форм, или типов, в настоящее время по предложенной в 1979 г. D.O. Sillence классификации несовершенный остеогенез с учетом клинических, рентгенологических и коллагеновых протеин-генных молекулярных изменений подразделяют на 4 типа.
Тип I — слабовыраженная форма, доминантно-наследственный несовершенный остеогенез с ломкостью костей и голубыми склерами.
Тип II — перинатально-летальный.
Тип III — прогрессирующее деформирование скелета.
Тип IV — доминантный с нормальными склерами и нерезко выраженными деформациями.
P.A. Dawson и др. (1999) выявили мутации типа I коллагеновых генов как причину всех четырех типов несовершенного остеогенеза (OI). У 2 детей рентгенограммы показали сниженную костную плотность поясничного отдела позвоночника и множественные переломы по всему позвоночнику; эта патология обусловлена изменениями в белках, особенно коллагена типа I. Энзимные изменения касалась единственной базальной мутации (1715 GA) у этих детей. Такая мутация предсказывает замену аргинина на глицин в позиции п43б (С4збК) в а2 (I), отец ребенка имел мутацию ДНК гена. Существование такой же гетерозиготной мутации у 2 детей предполагает, что пробанды отражают полностью этот фенотип. Клинические, биохимические и молекулярные находки расширяют представления о фенотипе, сочетанном с мутациями типа I коллагена, вызывающими изменения в позвоночнике, карликовость в подростковом возрасте.
На основании литературных публикаций последних лет, а также данных, приведенных на 3-й Международной конференции по несовершенному остеогенезу в 1985 г., и работ D.O. Sillence (1985) и др. приводим краткую характеристику этих 4 типов.
Тип I. Остеопороз и переломы костей чаще наблюдаются в раннем возрасте; после 10 лет частота их возникновения уменьшается и опять увеличивается после 40 лет. Переломы приводят к деформации костей. У 50 % больных отмечается небольшой рост. Голубизна склер усугубляется преждевременным появлением старческого ободка. У части больных дентин не изменен, тогда как у другой части его называют опаловым. Встречаются изменения аорты и митральный порок сердца, носовые кровотечения. У 20 % больных с НО I типа наблюдается пролапс митрального клапана. Такой больной описан И.А. Шамовым и Ш.М. Захарьевским в 1989 г. Эта форма обусловлена структурными мутациями в спиральном домене про-а,, возможность передачи по наследству около 7 %.
Тип II. Перинатально-летальный несовершенный остеогенез. Клинически и биохимически это гетерогенная группа больных, для которых характерны внутриутробная или ранняя неонатальная смерть, множественность и легкость наступления переломов. Подразделяется на три группы.
Группа А. Хрупкость соединительнотканных образований настолько выражена, что повреждения конечностей и головы плода происходят еще во время беременности; мозговой череп непропорционально велик, грудная клетка маленькая, конечности укорочены и искривлены, встречаются очень тяжелые степени обызвествления стенок аорты и эндокарда, очень малый рост при рождении (иногда 30—25 см).
Часто преждевременные роды: в 15 % случаев в ягодичном предлежа нии, до 20 % — мертворожденные, остальные умирают или в первые дни, или на 4-й неделе жизни. Рентгенологические изменения определяются у плода еще до рождения: широкие бедренные кости с волнистыми краями, короткая грудная клетка, ребра с четками и т.д. Согласно генетическим данным большинство таких случаев являются спорадическими. Биохимические данные позволяют предположить, что больные группы А «... являются гетерогенными для мутаций, вызывающих нарушение npo-oci (I) коллагеновых цепей, приводя к дефектной triple helical assembly секвестрации и инкорпорации внутрь нормальной соединительной ткани. Малое количество больных обладает гетерозиготными мутациями в npo-ai (I) коллагеновой цепи, в то время как некоторые другие описывались с единичной аминокислотной заменой, т.е. глицина на цистин, приводя к формированию дисульфатных мостиков между двумя цепями cti (I) и избыточному скоплению I типа коллагеновых молекул» [Sillence D.O., 1985]. Обследование пробандов говорит о возможном молекулярном дефекте, который совместим с гетерозиготностью мутаций в коллагеновом гене, что проявляется в особенностях наследования — аутосомно-доминантном.
Группа Б по фенотипу похожа на группу А, однако нарушения дыхательной системы менее выражены и больные живут несколько лет. Трубчатые кости укорочены и расширены, изменены ребра, но переломы их редки. Предполагается аутосомно-рецессивная наследственность вследствие свежей мутации.
Группа В наблюдается редко, часто отмечаются мертворожденность и смертность в течение первого месяца жизни. Больные маленького роста, трубчатые кости тонкие, особенно диафизы, отсутствует оссификация в костях мозгового и лицевого черепа. Предполагается аутосомно-рецессивная наследственность.
Тип III встречается относительно редко, тело новорожденных укорочено, масса тела может быть нормальной, переломы иногда происходят в процессе родов, а иногда в возрасте нескольких лет. Формируются деформации конечностей (О-образные), кифосколиоз, особенно прогрессирующие во время пубертатности. Изменения скелета и сердечно-сосудистой системы приводят к смерти 40—50 % больных. Резко выражен остеопороз — остеопения, нарушены оссификация и рост костей в длину, в ростковых зонах костей — неравномерное обызвествление, приводящее к образованию пятнистости («кукурузные зерна»).
Как указывает D.O. Sillence (1985), для этого типа характерна аутосомно-рецессивная наследственность. Только у одного больного он мог констатировать, что фенотип образовался благодаря гомозиготности для молекулярного дефекта в коллагене. Наследственность свежая аутосомная, доминантная мутация или аутосомно-рецессивная.
Тип IV. Изменения в скелете встречаются наиболее часто. Характерна большая вариабельность остеопении, возраста, количества переломов костей, голубизны склер (у взрослых склеры могут быть нормального цвета). Количество переломов с возрастом уменьшается, происходит нормальное образование костной мозоли, в возрасте старше 30 лет у V3 больных нарушается слух. Больные этого типа несовершенного остеогенеза подразделяются на две группы: с резко измененными опаловыми зубами и без изменений зубов. Преобладание аутосомно-доминантной наследственности выражается резко благодаря отсутствию фенотипического маркера (как голубые склеры).
В настоящее время считается, что несовершенный остеогенез обусловлен качественными и количественными изменениями в синтезе коллагена I типа. При I типе несовершенного остеогенеза синтез структурно нормального коллагена снижен, тогда как при II и IV типах синтез такого коллагена бывает нормальным, но из-за пониженной стабильности общее количество коллагена снижается. По данным D.O. Sillence (1985), число коллагеновых молекул, продуцируемых при несовершенном остеогенезе, быстро и постоянно увеличивается, но все же не достигает нормы. Поэтому он считает, что в данном случае наблюдается не простое нарушение синтеза коллагена вследствие изменения 4-й хромосомы, а нарушение свойств соединительной ткани, вызванное изменением и протеогликанового синтеза, и генаколлагена.
D.H. Colin и Р.Н. Byers (1991) обнаружили следующее: у 4 больных из 60 клеток синтезировали популяцию цепи а2 (I) с остатками цистина в тройной спирали, а клинические различия и гетерогенность в локализации остатков цистина дают основание предположить, что положение и места замены внутри самой цепи являются важными в определении клинического фенотипа. Этим подтверждается мнение о том, что больные с нелетальной формой несовершенного остеогенеза могут часто иметь дефекты в COL A1 или в COL 1A2 генах, предполагая, что многие из таких дефектов замещаются на остатки глицина в оа (I) тройного спирального пространства.
L. Cohen-Solal и др. (1991) показали, что тип II и тип III несовершенного остеогенеза могут появиться вследствие гонадного мозаицизма. что очень важно для генетической консультации при определении соответствующего фенотипа заболевания.
Анализы на проколлаген типа I молекул, синтезированных дермальными фибробластами, культивированными от больных с несовершенным остеогенезом, позволили установить две обширные биохимические группы: 1) больные, фибробласты которых синтезировали и эффективно секретировали около половины ожидаемого количества структурно нормального проколлагена I типа [Barsh G.S. et al., 1981; Genovese C., Rowe D.W., 1987; Willing M.C. et al., 1990]; 2) больные, фибробласты которых продуцировали нормальные и ненормальные популяции молекул и затем секретировали их [Bonadio J. et al., 1985; Wenstrup R.J. et al., 1986].
R.J. Wenstrup и др. (1990) сообщили, что они провели аналогичные исследования у 224 больных и сравнили полученные биохимические данные с клинической картиной. Оказалось, что в 1-й группе, где наблюдалось уменьшение количества нормального проколлагена типа I, клинические проявления были небольшими, а во 2-й группе, где обнаруживался синтез нормальных молекул и ненормального проколлагена типа I, фенотип варьировал от умеренно деформирующего кости и со слегка укороченной фигурой до заболевания, резко деформирующего скелет с умеренно или резко укороченной фигурой. Эти и другие исследования позволяют ставить пренатальный диагноза. По мнению R.J. Wenstup и др. (1990), при лечении нужно учитывать биохимические дефекты.
Л.М. Михайлова (1971) при ультрамикроскопическом исследовании костной ткани больных с несовершенным остеогенезом во многих остеобластах отметила редукцию элементов гранулярного эндоплазматического ретикулума, что вызывало нарушение фибриллогенеза; оказывались измененными также митохондрии, в матриксе которых имелись скопления кристаллов (очевидно, гидроксиапатита), что, по ее мнению, свидетельствовало о нарушении кальциевых и фосфатных ионов. По данным М.В. Волкова и Н.Н. Нефедьевой (1974), у больных резко увеличено содержание гексоз, гликопротеидов, гексозаминов, сиалопротеидов в сыворотке крови и с мочой выделяется повышенное количество мукополисахаридов. Патологические изменения у больных с несовершенным остеогенезом весьма разнообразны.
Псевдосаркомы. После перелома развивается костная мозоль больших пли громадных размеров (рис. 5.1), резко поротичная, постепенно, в течение ряда лет или десятилетий, увеличивающаяся, которую приходится дифференцировать от саркомы, тем более что в литературе имеются указания на развитие остеогенной саркомы у больных с НО. Развитие псевдосаркомы сопровождается довольно сильными болевыми ощущениями, напряжением тканей, местной гиперемией.
Развитие костной мозоли больших размеров, по мнению Т.П. Виноградовой (1973), является механизмом, компенсирующим недостаточную прочность ее структур. После срастания отломков эти опухолеподобные мозоли исчезают. Однако очень редко у больных с НО костные мозоли не рассасываются, а остаются необычно большими (какими были первоначально) или медленно продолжают расти, так что их уже невозможно принимать за проявление компенсаторного процесса. Удовлетворительных гипотез их происхождения нет. Мы наблюдали 3 больных с развитием «псевдосарком», у 2 из которых они достигали гигантских размеров.
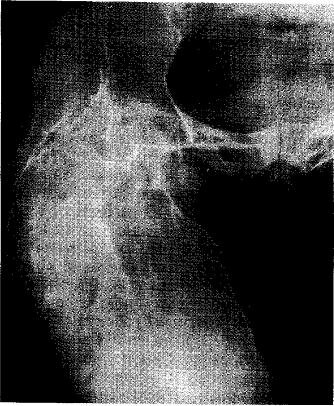
Рис. 5.1. Костная мозоль, обусловившая увеличение правой бедренной кости, — псевдосаркома.
Одну больную мы оперировали. Костная ткань имела вид спонгиозы с тонкими перегородками и большими лакунами жирового костного мозга.
Создалось впечатление, что разрастание костного мозга приводит к увеличению объема кости, костных лакун, а реактивное костеобразование способно только к образованию тонких перегородок и полостей, но не способно остановить процесс, в связи с чем не может образоваться нормальный кортикальный слой.
Мы считаем допустимым предположить, что при НО наблюдаемая остеопения является следствием, во-первых, некоторого уменьшения количества «активных ячеек роста костной ткани», которые, согласно теории, разработанной Н.М. Frost и др., определяют моделирование костной ткани; во-вторых, следствием изменений в коллагеновых структурах и, в-третьих, очевидно, следствием нарушений обмена в «третьей разновидности жировой ткани». По А.А. Заварзину (1985), такой разновидностью является жировая ткань костного мозга, жировые клетки которого содержат особые липиды, обычно не используемые в липидном обмене. Бурная пролиферация соединительной ткани, наблюдаемая при переломах и развитии псевдосаркомы [Fairbank H.A.T., Baker S.L., 1948], способствует образованию больших лакун и тем самым спонгизации кости: на участках, где развивается псевдосаркома, иногда кортикальный слой как таковой не определяется.
А.Н. Черняев и Г.А. Грибанов (1982) показали, что продолжительное введение кальцитонина способствует увеличению синтеза фибробластами не только коллагена, гликозаминогликанов, но и липидов. Естественно, необходимо тщательно исследовать в динамике уровень выработки кальцитонина у больных с псевдосаркоматозными формами несовершенного остеогенеза. Нам пришлось в течение 30 лет наблюдать больную с резко выраженной формой псевдосаркоматозной формы несовершенного остеогенеза. Он протекает не равномерно, а стадийно, период медленного спокойного течения сменяется периодом бурного развития, появляются боли в той или иной кости, местно повышается температура, что сопровождается появлением участков гиперемии без четких границ, резко возрастает уровень щелочной фосфатазы.
Больная А. наблюдалась нами с возраста 33 лет до 61 года. Родилась нормальным ребенком в 1933 г., самостоятельно ходила до 1 года 9 мес, когда произошел перелом правого бедра. Через год — повторный перелом правого бедра, в возрасте 6 лет — перелом костей правой голени, затем левой бедренной кости, всего было 7 переломов. Больную консультировали известные специалисты: Г.С. Бом, П.А. Герцен (сказал — «проживет не больше года»), С.М. Спасокукоцкий, Т.П. Краснобаев («у этой болезни нет названия»), И.Г. Лагунова, М.К. Климова. В 1970 г. обратилась в ЦИТО и была стационирована с диагнозом: несовершенный остеогенез, псевдосаркоматозная форма.
Больная очень маленького роста (107 см), с трудом ходит на костылях, предпочитает передвигаться на каталке. Жалобы на постоянно увеличивающееся в объеме правое бедро, представлявшее собой несколько вытянутый «арбуз», вверху переходивший в таз, а внизу заканчивавшийся у колена. Были увеличены в объеме также большеберцовая кость и левое бедро. Движений в правом тазобедренном суставе практически не было, и больная не могла произвести туалет промежности, а при мочеиспускании моча попадала на внутреннюю поверхность бедра. Нами произведена подвертельная остеотомия правой бедренной кости, при этом понадобился не молоток, а долото, которое под нажимом руки легко погружалось в кость, представлявшую жировой костный мозг, разделенный тонкими костными перегородками. Произведена остеотомия 3 /4 поперечника бедренной кости, после чего нога отведена кнаружи и фиксирована гипсовой лонгетой. Клинически патологическая измененная кость производила впечатление разрастающегося костномозгового жира и остеопорозной истонченной костной ткани: редкие атрофичные костные трабекулы.
В течение 25 лет значительных перемен в состоянии больной не было. В 1995 г. произошел перелом бедренной кости, после чего ее объем стал быстро увеличиваться, как и объем левой голени, больная с трудом переворачивалась на кровати. При осмотре в 1997 г. оба бедра и голени резко увеличены в объеме. Увеличены и все кости таза с обеих сторон, состояние больной тяжелое. Через месяц по телефону мне сообщили, что у нее произошел перелом нескольких ребер, собираются положить в больницу. Связь прервалась.
Лечение. В настоящее время принято считать, что при всех формах НО показано лечение остеопороза витамином D3, комплексонами (ксидифоном и др.), бифосфонатами, глюконатом кальция, глицерофосфатом, солями магния, калия. Реже применялось лечение рыбьим жиром, витамином D2, анаболическими гормонами, ультрафиолетовым облучением [Волков М.Б., Нефедьева Н.Н., 1974]. Большее распространение и эффект давало лечение, разработанное в 1984 г. Н.А. Беловой в виде схемы и рассчитанное на 12 мес (соматотропный гормон по 4 ЕД 3 раза в неделю в течение 1-го и 9-го месяцев; кальцитрин по 3—7,5 ЕД ежедневно в течение 2-го и 10-го месяцев; витамин D2 — 9-й и 12-й месяцы; оксидевит (витамин D3) по 1 — 1,5 мкг в сутки — 3-й, 4-й и 11-й, 12-й месяцы; фестал, панзинорм, глюконат кальция, фитин, цитратная смесь, витамины А, Е, электрофорез с солями кальция, массаж, ЛФК). По данным А.П. Бережного с соавт. (1988), это консервативное лечение позволило получить положительные результаты: у ряда больных прекратились переломы длинных трубчатых костей, а проведенное в предоперационном периоде лечение позволило улучшить результаты операций. Таким образом, консервативное лечение с применением витамина D3 и других препаратов следует проводить всем больным с НО.
Консервативное лечение переломов костей у этой группы больных является довольно сложной задачей, поскольку у некоторых из них переломы возникают часто, а иногда бывают множественными. Необходимо использовать все имеющиеся методы лечения, а иногда ставить показания к оперативному вмешательству.
Учитывая повышенную ломкость костей, некоторые ортопеды для исправления деформации осуществляли остеоклазию на вершине искривления, исправляли деформацию и фиксировали конечность гипсовой повязкой или вытяжением.
Оперативное лечение в 40—50-х годах осуществлялось у единичных больных. Ф.Р. Богданов (1945) производил сегментарные остеотомии, а для интрамедуллярной фиксации применял предложенный им штифт. Т.С. Зацепин использовал штифты из гетерокости и металла. В 1964 г. М.В. Волков предложил в качестве интрамедуллярного фиксатора аллогенные трансплантаты, а затем разработал методику, которая включает декортикацию деформированной кости, сегментарную остеотомию и пластику с помощью аллотрансплантатов по типу «вязанки хвороста». Эта методика оказалась очень эффективной, аллогенные трансплантаты при этом спаиваются остеогенной тканью и постепенно перестраиваются.
В руководимом нами отделении оперативное лечение произведено 43 таким больных, которым в общей сложности выполнено 91 оперативное вмешательство. Ортопедам, занимающимся оперативным лечением больных с НО, приходится учитывать изменения скелета у больного и в зависимости от этого ставить хирургические задачи, вырабатывать план и выбирать методы лечения. Мы наблюдали разные клинические формы и предлагаем их подразделять на следующие группы.
С.Т.Зацепин
Костная патология взрослых
Lobstein (1833) описал ломкость костей у больных различного возраста. По данным Vrolik (1849), переломы у детей происходили или еще внутриутробно, или вскоре после рождения. Е. Looser (1906) описал эти две формы как osteogenesis imperfecta congenita und tarda.
Изучением заболевания занимались многие врачи, описавшие более 20 различных симптомов, из которых основными являются:
изменения в строении скелета и легко наступающие переломы, часто небольшой рост; голубые склеры; опаловидный дентин (dentinogenesis imperfecta); прогрессирующая деформация позвоночника, грудной клетки, черепа и длинных трубчатых костей; тугоухость по проводниковому типу; гиперэкстензия в суставах и их деформация; изменения со стороны сердца и крупных сосудов, носовые кровотечения и др.
Работами последних лет показано, что несовершенный остеогенез является гетерогенным наследственным заболеванием генетической природы, поражающим соединительную ткань и выражающимся остеопенией и вышеперечисленными клиническими признаками.
Вместо двух форм, или типов, в настоящее время по предложенной в 1979 г. D.O. Sillence классификации несовершенный остеогенез с учетом клинических, рентгенологических и коллагеновых протеин-генных молекулярных изменений подразделяют на 4 типа.
Тип I — слабовыраженная форма, доминантно-наследственный несовершенный остеогенез с ломкостью костей и голубыми склерами.
Тип II — перинатально-летальный.
Тип III — прогрессирующее деформирование скелета.
Тип IV — доминантный с нормальными склерами и нерезко выраженными деформациями.
P.A. Dawson и др. (1999) выявили мутации типа I коллагеновых генов как причину всех четырех типов несовершенного остеогенеза (OI). У 2 детей рентгенограммы показали сниженную костную плотность поясничного отдела позвоночника и множественные переломы по всему позвоночнику; эта патология обусловлена изменениями в белках, особенно коллагена типа I. Энзимные изменения касалась единственной базальной мутации (1715 GA) у этих детей. Такая мутация предсказывает замену аргинина на глицин в позиции п43б (С4збК) в а2 (I), отец ребенка имел мутацию ДНК гена. Существование такой же гетерозиготной мутации у 2 детей предполагает, что пробанды отражают полностью этот фенотип. Клинические, биохимические и молекулярные находки расширяют представления о фенотипе, сочетанном с мутациями типа I коллагена, вызывающими изменения в позвоночнике, карликовость в подростковом возрасте.
На основании литературных публикаций последних лет, а также данных, приведенных на 3-й Международной конференции по несовершенному остеогенезу в 1985 г., и работ D.O. Sillence (1985) и др. приводим краткую характеристику этих 4 типов.
Тип I. Остеопороз и переломы костей чаще наблюдаются в раннем возрасте; после 10 лет частота их возникновения уменьшается и опять увеличивается после 40 лет. Переломы приводят к деформации костей. У 50 % больных отмечается небольшой рост. Голубизна склер усугубляется преждевременным появлением старческого ободка. У части больных дентин не изменен, тогда как у другой части его называют опаловым. Встречаются изменения аорты и митральный порок сердца, носовые кровотечения. У 20 % больных с НО I типа наблюдается пролапс митрального клапана. Такой больной описан И.А. Шамовым и Ш.М. Захарьевским в 1989 г. Эта форма обусловлена структурными мутациями в спиральном домене про-а,, возможность передачи по наследству около 7 %.
Тип II. Перинатально-летальный несовершенный остеогенез. Клинически и биохимически это гетерогенная группа больных, для которых характерны внутриутробная или ранняя неонатальная смерть, множественность и легкость наступления переломов. Подразделяется на три группы.
Группа А. Хрупкость соединительнотканных образований настолько выражена, что повреждения конечностей и головы плода происходят еще во время беременности; мозговой череп непропорционально велик, грудная клетка маленькая, конечности укорочены и искривлены, встречаются очень тяжелые степени обызвествления стенок аорты и эндокарда, очень малый рост при рождении (иногда 30—25 см).
Часто преждевременные роды: в 15 % случаев в ягодичном предлежа нии, до 20 % — мертворожденные, остальные умирают или в первые дни, или на 4-й неделе жизни. Рентгенологические изменения определяются у плода еще до рождения: широкие бедренные кости с волнистыми краями, короткая грудная клетка, ребра с четками и т.д. Согласно генетическим данным большинство таких случаев являются спорадическими. Биохимические данные позволяют предположить, что больные группы А «... являются гетерогенными для мутаций, вызывающих нарушение npo-oci (I) коллагеновых цепей, приводя к дефектной triple helical assembly секвестрации и инкорпорации внутрь нормальной соединительной ткани. Малое количество больных обладает гетерозиготными мутациями в npo-ai (I) коллагеновой цепи, в то время как некоторые другие описывались с единичной аминокислотной заменой, т.е. глицина на цистин, приводя к формированию дисульфатных мостиков между двумя цепями cti (I) и избыточному скоплению I типа коллагеновых молекул» [Sillence D.O., 1985]. Обследование пробандов говорит о возможном молекулярном дефекте, который совместим с гетерозиготностью мутаций в коллагеновом гене, что проявляется в особенностях наследования — аутосомно-доминантном.
Группа Б по фенотипу похожа на группу А, однако нарушения дыхательной системы менее выражены и больные живут несколько лет. Трубчатые кости укорочены и расширены, изменены ребра, но переломы их редки. Предполагается аутосомно-рецессивная наследственность вследствие свежей мутации.
Группа В наблюдается редко, часто отмечаются мертворожденность и смертность в течение первого месяца жизни. Больные маленького роста, трубчатые кости тонкие, особенно диафизы, отсутствует оссификация в костях мозгового и лицевого черепа. Предполагается аутосомно-рецессивная наследственность.
Тип III встречается относительно редко, тело новорожденных укорочено, масса тела может быть нормальной, переломы иногда происходят в процессе родов, а иногда в возрасте нескольких лет. Формируются деформации конечностей (О-образные), кифосколиоз, особенно прогрессирующие во время пубертатности. Изменения скелета и сердечно-сосудистой системы приводят к смерти 40—50 % больных. Резко выражен остеопороз — остеопения, нарушены оссификация и рост костей в длину, в ростковых зонах костей — неравномерное обызвествление, приводящее к образованию пятнистости («кукурузные зерна»).
Как указывает D.O. Sillence (1985), для этого типа характерна аутосомно-рецессивная наследственность. Только у одного больного он мог констатировать, что фенотип образовался благодаря гомозиготности для молекулярного дефекта в коллагене. Наследственность свежая аутосомная, доминантная мутация или аутосомно-рецессивная.
Тип IV. Изменения в скелете встречаются наиболее часто. Характерна большая вариабельность остеопении, возраста, количества переломов костей, голубизны склер (у взрослых склеры могут быть нормального цвета). Количество переломов с возрастом уменьшается, происходит нормальное образование костной мозоли, в возрасте старше 30 лет у V3 больных нарушается слух. Больные этого типа несовершенного остеогенеза подразделяются на две группы: с резко измененными опаловыми зубами и без изменений зубов. Преобладание аутосомно-доминантной наследственности выражается резко благодаря отсутствию фенотипического маркера (как голубые склеры).
В настоящее время считается, что несовершенный остеогенез обусловлен качественными и количественными изменениями в синтезе коллагена I типа. При I типе несовершенного остеогенеза синтез структурно нормального коллагена снижен, тогда как при II и IV типах синтез такого коллагена бывает нормальным, но из-за пониженной стабильности общее количество коллагена снижается. По данным D.O. Sillence (1985), число коллагеновых молекул, продуцируемых при несовершенном остеогенезе, быстро и постоянно увеличивается, но все же не достигает нормы. Поэтому он считает, что в данном случае наблюдается не простое нарушение синтеза коллагена вследствие изменения 4-й хромосомы, а нарушение свойств соединительной ткани, вызванное изменением и протеогликанового синтеза, и генаколлагена.
D.H. Colin и Р.Н. Byers (1991) обнаружили следующее: у 4 больных из 60 клеток синтезировали популяцию цепи а2 (I) с остатками цистина в тройной спирали, а клинические различия и гетерогенность в локализации остатков цистина дают основание предположить, что положение и места замены внутри самой цепи являются важными в определении клинического фенотипа. Этим подтверждается мнение о том, что больные с нелетальной формой несовершенного остеогенеза могут часто иметь дефекты в COL A1 или в COL 1A2 генах, предполагая, что многие из таких дефектов замещаются на остатки глицина в оа (I) тройного спирального пространства.
L. Cohen-Solal и др. (1991) показали, что тип II и тип III несовершенного остеогенеза могут появиться вследствие гонадного мозаицизма. что очень важно для генетической консультации при определении соответствующего фенотипа заболевания.
Анализы на проколлаген типа I молекул, синтезированных дермальными фибробластами, культивированными от больных с несовершенным остеогенезом, позволили установить две обширные биохимические группы: 1) больные, фибробласты которых синтезировали и эффективно секретировали около половины ожидаемого количества структурно нормального проколлагена I типа [Barsh G.S. et al., 1981; Genovese C., Rowe D.W., 1987; Willing M.C. et al., 1990]; 2) больные, фибробласты которых продуцировали нормальные и ненормальные популяции молекул и затем секретировали их [Bonadio J. et al., 1985; Wenstrup R.J. et al., 1986].
R.J. Wenstrup и др. (1990) сообщили, что они провели аналогичные исследования у 224 больных и сравнили полученные биохимические данные с клинической картиной. Оказалось, что в 1-й группе, где наблюдалось уменьшение количества нормального проколлагена типа I, клинические проявления были небольшими, а во 2-й группе, где обнаруживался синтез нормальных молекул и ненормального проколлагена типа I, фенотип варьировал от умеренно деформирующего кости и со слегка укороченной фигурой до заболевания, резко деформирующего скелет с умеренно или резко укороченной фигурой. Эти и другие исследования позволяют ставить пренатальный диагноза. По мнению R.J. Wenstup и др. (1990), при лечении нужно учитывать биохимические дефекты.
Л.М. Михайлова (1971) при ультрамикроскопическом исследовании костной ткани больных с несовершенным остеогенезом во многих остеобластах отметила редукцию элементов гранулярного эндоплазматического ретикулума, что вызывало нарушение фибриллогенеза; оказывались измененными также митохондрии, в матриксе которых имелись скопления кристаллов (очевидно, гидроксиапатита), что, по ее мнению, свидетельствовало о нарушении кальциевых и фосфатных ионов. По данным М.В. Волкова и Н.Н. Нефедьевой (1974), у больных резко увеличено содержание гексоз, гликопротеидов, гексозаминов, сиалопротеидов в сыворотке крови и с мочой выделяется повышенное количество мукополисахаридов. Патологические изменения у больных с несовершенным остеогенезом весьма разнообразны.
Псевдосаркомы. После перелома развивается костная мозоль больших пли громадных размеров (рис. 5.1), резко поротичная, постепенно, в течение ряда лет или десятилетий, увеличивающаяся, которую приходится дифференцировать от саркомы, тем более что в литературе имеются указания на развитие остеогенной саркомы у больных с НО. Развитие псевдосаркомы сопровождается довольно сильными болевыми ощущениями, напряжением тканей, местной гиперемией.
Развитие костной мозоли больших размеров, по мнению Т.П. Виноградовой (1973), является механизмом, компенсирующим недостаточную прочность ее структур. После срастания отломков эти опухолеподобные мозоли исчезают. Однако очень редко у больных с НО костные мозоли не рассасываются, а остаются необычно большими (какими были первоначально) или медленно продолжают расти, так что их уже невозможно принимать за проявление компенсаторного процесса. Удовлетворительных гипотез их происхождения нет. Мы наблюдали 3 больных с развитием «псевдосарком», у 2 из которых они достигали гигантских размеров.
Рис. 5.1. Костная мозоль, обусловившая увеличение правой бедренной кости, — псевдосаркома.
Одну больную мы оперировали. Костная ткань имела вид спонгиозы с тонкими перегородками и большими лакунами жирового костного мозга.
Создалось впечатление, что разрастание костного мозга приводит к увеличению объема кости, костных лакун, а реактивное костеобразование способно только к образованию тонких перегородок и полостей, но не способно остановить процесс, в связи с чем не может образоваться нормальный кортикальный слой.
Мы считаем допустимым предположить, что при НО наблюдаемая остеопения является следствием, во-первых, некоторого уменьшения количества «активных ячеек роста костной ткани», которые, согласно теории, разработанной Н.М. Frost и др., определяют моделирование костной ткани; во-вторых, следствием изменений в коллагеновых структурах и, в-третьих, очевидно, следствием нарушений обмена в «третьей разновидности жировой ткани». По А.А. Заварзину (1985), такой разновидностью является жировая ткань костного мозга, жировые клетки которого содержат особые липиды, обычно не используемые в липидном обмене. Бурная пролиферация соединительной ткани, наблюдаемая при переломах и развитии псевдосаркомы [Fairbank H.A.T., Baker S.L., 1948], способствует образованию больших лакун и тем самым спонгизации кости: на участках, где развивается псевдосаркома, иногда кортикальный слой как таковой не определяется.
А.Н. Черняев и Г.А. Грибанов (1982) показали, что продолжительное введение кальцитонина способствует увеличению синтеза фибробластами не только коллагена, гликозаминогликанов, но и липидов. Естественно, необходимо тщательно исследовать в динамике уровень выработки кальцитонина у больных с псевдосаркоматозными формами несовершенного остеогенеза. Нам пришлось в течение 30 лет наблюдать больную с резко выраженной формой псевдосаркоматозной формы несовершенного остеогенеза. Он протекает не равномерно, а стадийно, период медленного спокойного течения сменяется периодом бурного развития, появляются боли в той или иной кости, местно повышается температура, что сопровождается появлением участков гиперемии без четких границ, резко возрастает уровень щелочной фосфатазы.
Больная А. наблюдалась нами с возраста 33 лет до 61 года. Родилась нормальным ребенком в 1933 г., самостоятельно ходила до 1 года 9 мес, когда произошел перелом правого бедра. Через год — повторный перелом правого бедра, в возрасте 6 лет — перелом костей правой голени, затем левой бедренной кости, всего было 7 переломов. Больную консультировали известные специалисты: Г.С. Бом, П.А. Герцен (сказал — «проживет не больше года»), С.М. Спасокукоцкий, Т.П. Краснобаев («у этой болезни нет названия»), И.Г. Лагунова, М.К. Климова. В 1970 г. обратилась в ЦИТО и была стационирована с диагнозом: несовершенный остеогенез, псевдосаркоматозная форма.
Больная очень маленького роста (107 см), с трудом ходит на костылях, предпочитает передвигаться на каталке. Жалобы на постоянно увеличивающееся в объеме правое бедро, представлявшее собой несколько вытянутый «арбуз», вверху переходивший в таз, а внизу заканчивавшийся у колена. Были увеличены в объеме также большеберцовая кость и левое бедро. Движений в правом тазобедренном суставе практически не было, и больная не могла произвести туалет промежности, а при мочеиспускании моча попадала на внутреннюю поверхность бедра. Нами произведена подвертельная остеотомия правой бедренной кости, при этом понадобился не молоток, а долото, которое под нажимом руки легко погружалось в кость, представлявшую жировой костный мозг, разделенный тонкими костными перегородками. Произведена остеотомия 3 /4 поперечника бедренной кости, после чего нога отведена кнаружи и фиксирована гипсовой лонгетой. Клинически патологическая измененная кость производила впечатление разрастающегося костномозгового жира и остеопорозной истонченной костной ткани: редкие атрофичные костные трабекулы.
В течение 25 лет значительных перемен в состоянии больной не было. В 1995 г. произошел перелом бедренной кости, после чего ее объем стал быстро увеличиваться, как и объем левой голени, больная с трудом переворачивалась на кровати. При осмотре в 1997 г. оба бедра и голени резко увеличены в объеме. Увеличены и все кости таза с обеих сторон, состояние больной тяжелое. Через месяц по телефону мне сообщили, что у нее произошел перелом нескольких ребер, собираются положить в больницу. Связь прервалась.
Лечение. В настоящее время принято считать, что при всех формах НО показано лечение остеопороза витамином D3, комплексонами (ксидифоном и др.), бифосфонатами, глюконатом кальция, глицерофосфатом, солями магния, калия. Реже применялось лечение рыбьим жиром, витамином D2, анаболическими гормонами, ультрафиолетовым облучением [Волков М.Б., Нефедьева Н.Н., 1974]. Большее распространение и эффект давало лечение, разработанное в 1984 г. Н.А. Беловой в виде схемы и рассчитанное на 12 мес (соматотропный гормон по 4 ЕД 3 раза в неделю в течение 1-го и 9-го месяцев; кальцитрин по 3—7,5 ЕД ежедневно в течение 2-го и 10-го месяцев; витамин D2 — 9-й и 12-й месяцы; оксидевит (витамин D3) по 1 — 1,5 мкг в сутки — 3-й, 4-й и 11-й, 12-й месяцы; фестал, панзинорм, глюконат кальция, фитин, цитратная смесь, витамины А, Е, электрофорез с солями кальция, массаж, ЛФК). По данным А.П. Бережного с соавт. (1988), это консервативное лечение позволило получить положительные результаты: у ряда больных прекратились переломы длинных трубчатых костей, а проведенное в предоперационном периоде лечение позволило улучшить результаты операций. Таким образом, консервативное лечение с применением витамина D3 и других препаратов следует проводить всем больным с НО.
Консервативное лечение переломов костей у этой группы больных является довольно сложной задачей, поскольку у некоторых из них переломы возникают часто, а иногда бывают множественными. Необходимо использовать все имеющиеся методы лечения, а иногда ставить показания к оперативному вмешательству.
Учитывая повышенную ломкость костей, некоторые ортопеды для исправления деформации осуществляли остеоклазию на вершине искривления, исправляли деформацию и фиксировали конечность гипсовой повязкой или вытяжением.
Оперативное лечение в 40—50-х годах осуществлялось у единичных больных. Ф.Р. Богданов (1945) производил сегментарные остеотомии, а для интрамедуллярной фиксации применял предложенный им штифт. Т.С. Зацепин использовал штифты из гетерокости и металла. В 1964 г. М.В. Волков предложил в качестве интрамедуллярного фиксатора аллогенные трансплантаты, а затем разработал методику, которая включает декортикацию деформированной кости, сегментарную остеотомию и пластику с помощью аллотрансплантатов по типу «вязанки хвороста». Эта методика оказалась очень эффективной, аллогенные трансплантаты при этом спаиваются остеогенной тканью и постепенно перестраиваются.
В руководимом нами отделении оперативное лечение произведено 43 таким больных, которым в общей сложности выполнено 91 оперативное вмешательство. Ортопедам, занимающимся оперативным лечением больных с НО, приходится учитывать изменения скелета у больного и в зависимости от этого ставить хирургические задачи, вырабатывать план и выбирать методы лечения. Мы наблюдали разные клинические формы и предлагаем их подразделять на следующие группы.
С.Т.Зацепин
Костная патология взрослых
Комментировать:
Похожие статьи:
Редкие обменные заболевания костей
Категории: Костная патология, Метаболические заболевания костей,
Болезнь Кашина—Бека Болезнь Кашина—Бека (уровская болезнь) впервые была описана жителем города Нерченска И.М. Юренским в 1849 г. в «Трудах вольного экономического общества» под названием «Об уродливости..
Некоторые формы акроостеолиза и самопроизвольное исчезновение костей
Категории: Костная патология, Метаболические заболевания костей,
Работами большого числа исследователей было показано, что существует целый ряд заболеваний, обусловленных врожденным дефектом какого-то гена — «первичный молекулярный дефект». Это выявляется как нарушение..
Удаление гигантских доброкачественных и некоторых злокачественных опухолей крестца
Категории: Костная патология, Лечение при опухолях костей,
Доброкачественные опухоли крестца, особенно неврогенного происхождения — неврилеммомы, нейрофибромы, достигают иногда очень больших, даже гигантских размеров. Они смещают кверху и деформируют мочевой..
Удаление опухолей крестца
Категории: Костная патология, Лечение при опухолях костей,
Рис. 43.14. Тератома крестца. а разрушены III, IV, V крестцовые позвонки — тератома удалена. Метастазы рака в крестце выявлены у 72 пациентов (или 8 %) из 908 оперированных с метастатическими..